
Introduction
The binneR package provides a spectral binning approach for routine processing of flow infusion electrospray - high resolution mass spectrometry (FIE-HRMS) metabolomic fingerprinting experiments, the results of which can then be used for subsequent statistical analyses.
Spectral binning rounds high resolution fingerprinting data by a specified amu bin width. FIE-HRMS data consists of a ‘plug flow’, across which MS signal intensities can be averaged to provide a metabolome fingerprint. Below shows an animation of the spectrum change across ‘plug flow’ region of an example FIE-HRMS injection acquired in negative ionisation mode.
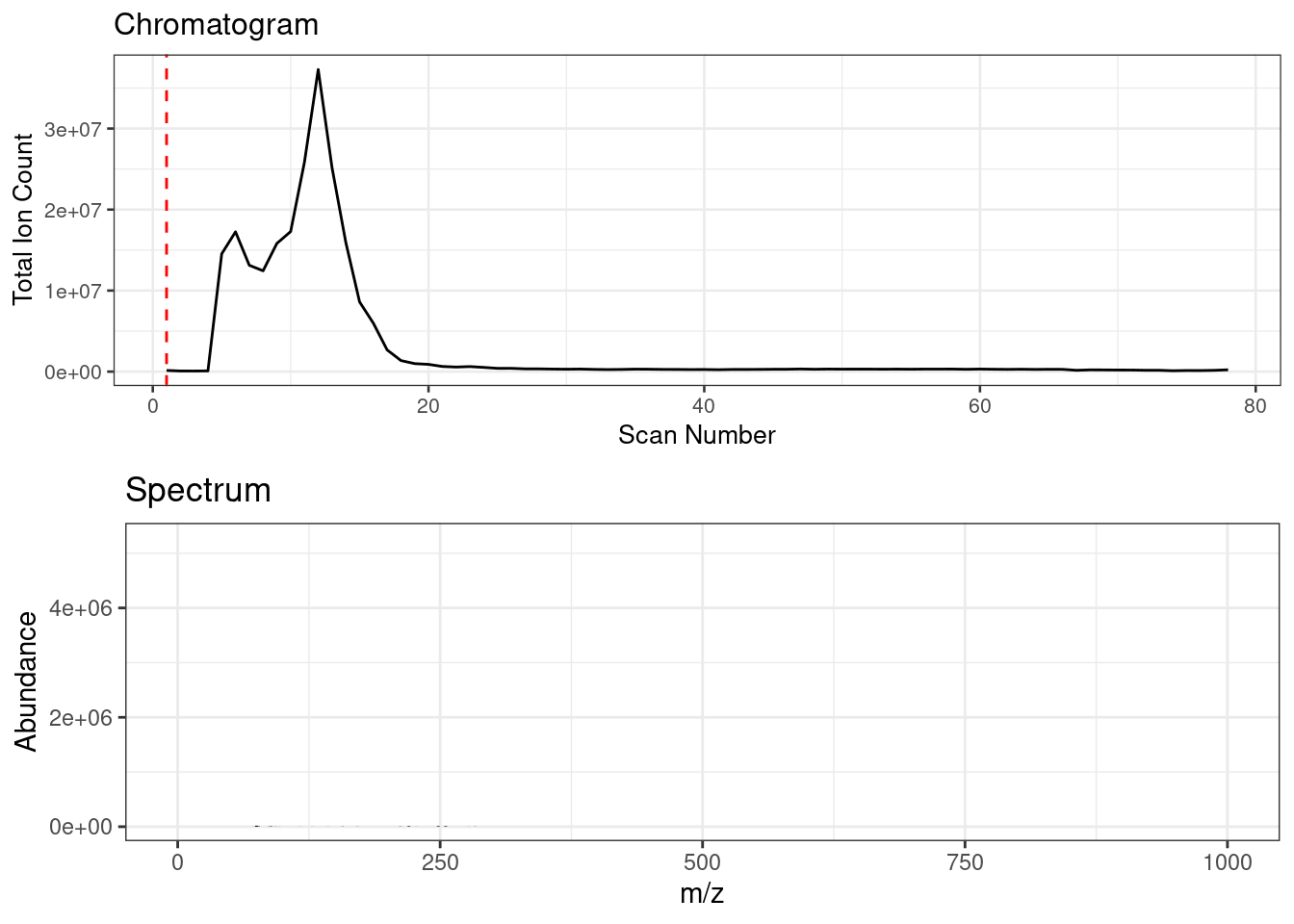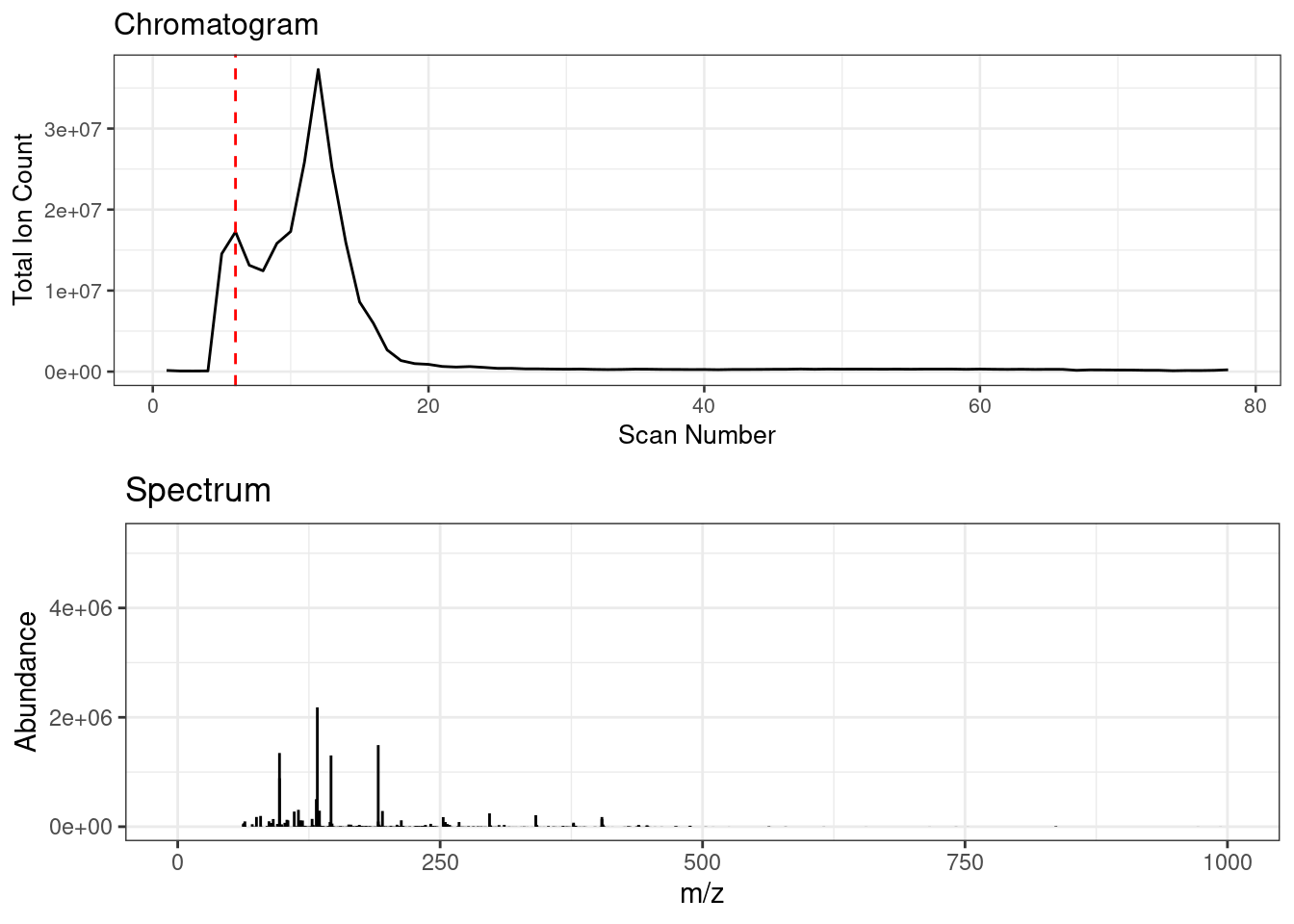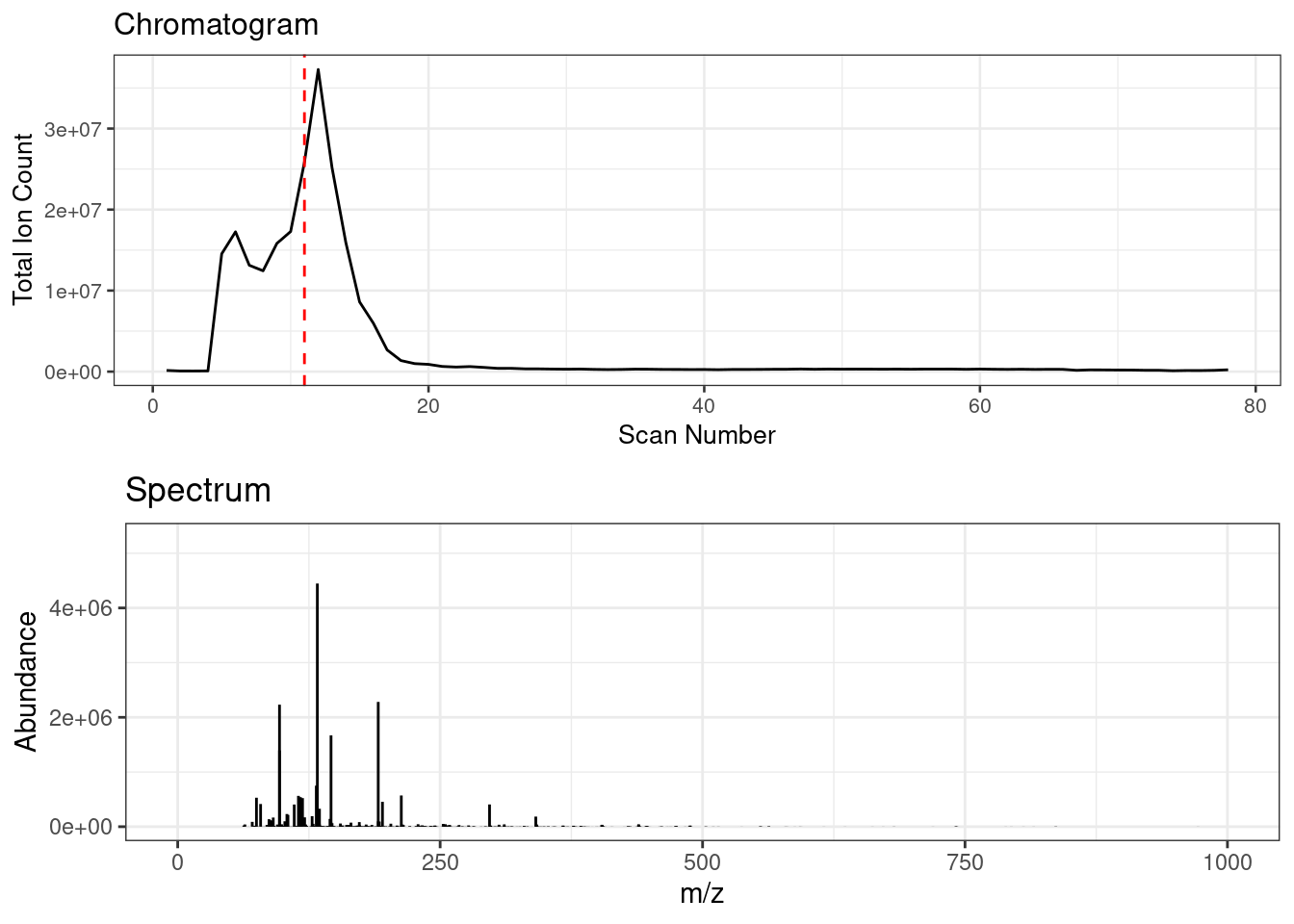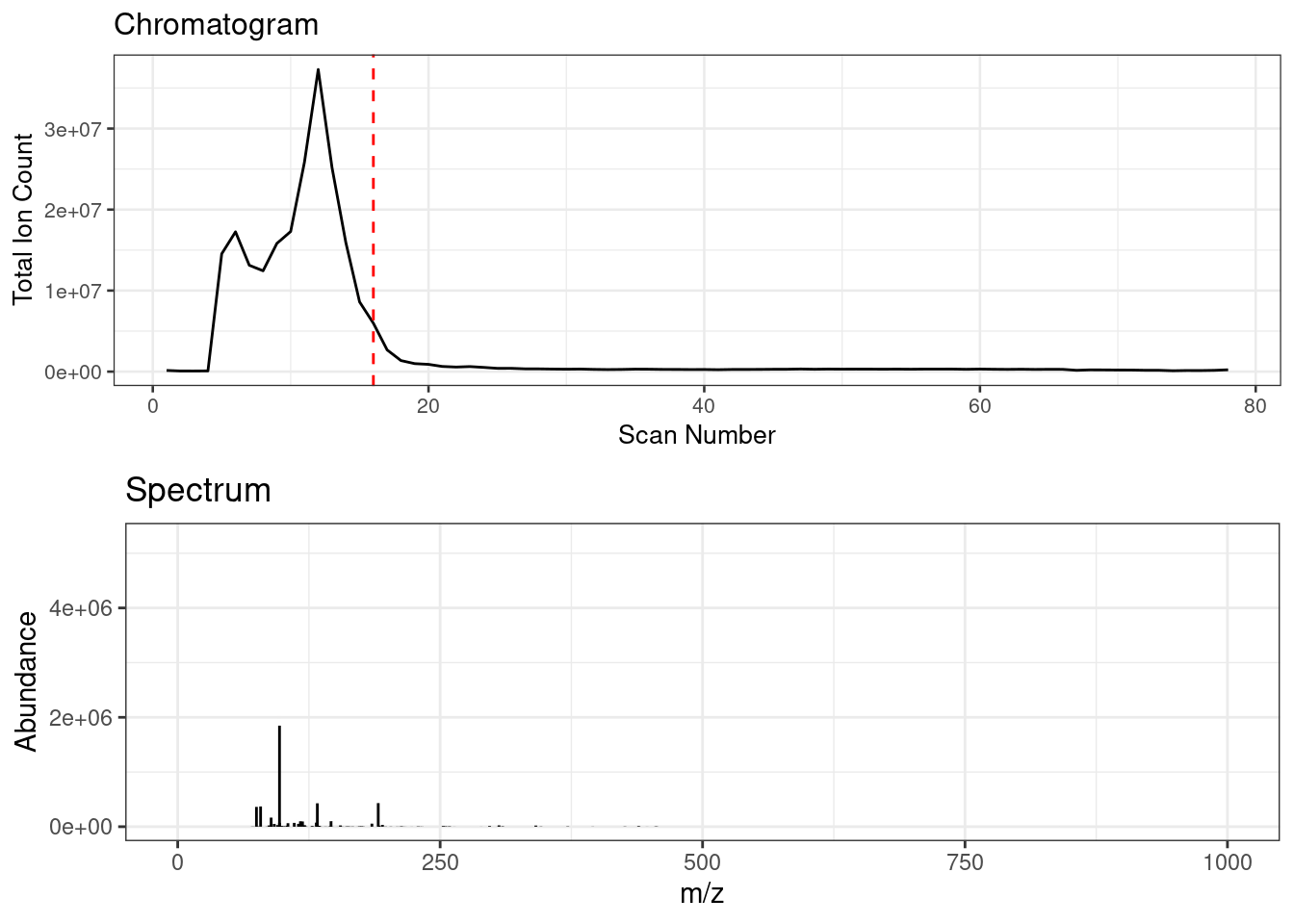
Spectral binning is applied on a scan by scan basis where the data is rounded to the specified bin width, the signals are then sum aggregated and their intensities are averaged across the specified scans.
Prior to the use of binneR, vendor specific raw data files need to be converted to one of the open source file formats such as .mzXML or .mzML so that they can be parsed into R. Data should also be centroided to reduce bin splitting artefacts that profile data can introduce during spectral binning. The msconvert tool can be used for both data conversion and centroiding, allowing the use of vendor specific algorithms.
There are two main functionalities provided by this package.
- Simple intensity matrix production - quick FIE-HRMS matrix investigations.
- binneRlyse - processing for routine metabolomics fingerprinting experiments.
The subsequent sections will outline the use of these two main functionalities.
Before we begin, the necessary packages need to be loaded.
Parallel Processing
The package supports parallel processing using the future package.
By default processing by binneR will be done
sequentially. However, parallel processing can be activated prior to
processing by specifying a parallel back-end using plan().
The following example specifies using the multisession
back-end (multiple background R sessions) with two worker processes.
plan(future::multisession,workers = 2)See the future package documentation for more information on the types of parallel back-ends that are available.
Infusion Scan Detection
In order to apply the spectral binning approach for FIE-HRMS data, the infusion scans need to be detected. For a set of specified file paths, the range of infusion scans can be detected using the following:
infusionScans <- detectInfusionScans(
metaboData::filePaths('FIE-HRMS','BdistachyonTechnical')[1],
thresh = 0.5
)The detected scans can then be checked by plotting an averaged chromatogram for these files. The infusion scans can also be plotted by supplying the range to the scans argument.
plotChromFromFile(
metaboData::filePaths('FIE-HRMS','BdistachyonTechnical')[1],
scans = infusionScans
)Simple Intensity Matrix Production - quick FIE-HRMS matrix investigations
The simplest funtionality of binneR is to read raw data
vector of specified file paths, bin these to a given amu and aggregate
across a given scan window. This can be useful for a quick assessment of
FIE-HRMS data structures. Spectral binning can be performed using the
readFiles() function as shown below. The example file
within the package can be specified using the following.
file <- metaboData::filePaths('FIE-HRMS','BdistachyonTechnical')[1]Then the data can be spectrally binned using:
res <- readFiles(file,dp = 2,scans = infusionScans)This will return a list containing the intensity matrices for each ionisation mode, with the rows being the individual samples and columns the spectral bins.
binneRlyse - metabolomics fingerprinting experiments
Routine FIE-HRMS metabolomic fingerprinting experiments can require
rapid the processing of hundereds of MS files that will also require
sample information such as biological classes for subsequent statistical
analyses. The package allows for a Binalysis that
formalises the spectral binning approach using an S4 class that not only
bins the data to 0.01 amu but will also extract accurate m/z
for each of these bins based on 0.00001 amu binned data. The accurate
m/z data can be aggregated based on a specified class structure
from which the modal accurate m/z is extracted. Some bin
metrics are also computed that allow the assessment of the quality of
the 0.01 amu bins.
The example data used here is from the metaboData package and consists of 10 replicate injections of a leaf tissue extract from the model grass species Brachypodium distachyon.
Basic Usage
Firstly the file paths and sample information can be loaded for the example data set using the following:
info <- metaboData::runinfo('FIE-HRMS','BdistachyonTechnical')
files <- metaboData::filePaths('FIE-HRMS','BdistachyonTechnical')There are two main functions for processing experimental data:
-
detectParameters()- allows the auto detection of processing parameters.binParameters()can be use to manually dplyr::select these parameters. -
binneRlyse()- input data file paths and sample information to process using the selected parameters.
Sample information
binneRlyse() requires the provision of sample
information (info) for the experimental run to be processed. This should
be in csv format and the recommended column headers
include:
-
fileOrder - the file order in alphabetical order as
returned by
list.files()
- injOrder - the injection order of the samples during FIE-HRMS analysis
- fileName - the sample file name
- batch - the sample batch
- block - the randomised block of the sample
- name - the sample name
- class - the sample class
The row orders of the info file should match the order in which the
files paths are submitted to the binneRlyse() processing
function.
Parameters
Prior to spectral binning the processing parameters first need to be
selected. The binning parameter can be detected using the
detectParameters() function as shown below.
parameters <- detectParameters(files)These parameters specify the following:
-
scans- the scan indexes to use for binning -
cls- the column of the info that contains class information if relevant
Alternatively, parameters can be initialised using the
binParameters function as shown below.
parameters <- binParameters(scans = 6:14)For and already initialised BinParameters object,
parameters can be changed using the methods named after the parameter of
interest. For example to change the scans of a given object:
alternative_parameters <- parameters
scans(alternative_parameters) <- 6:14Processing
Processing is simple and requires only the use of the
binneRlyse() function. The input of this function is a
vector of the paths of the data files to process, a tibble::tibble
containing the sample info and a BinParameters object.
It is crucial that the positions of the sample information in the info file match the sample positions within the files vector. Below shows an example of how this can be checked by matching the file names present in the info with those in the vector.
Spectral binning can then be performed with the following.
analysis <- binneRlyse(files,info,parameters)For data quality inspection, the infusion profiles this data can be plotted using:
plotChromatogram(analysis)The spectrum fingerprints using:
plotFingerprint(analysis)And the total ion counts using:
plotTIC(analysis)Density profiles for individual bins can be plotted by:
plotBin(analysis,'n133.01',type = 'cls')Data Extraction
There are a number of functions that can be used to return processing
data from a Binalysis object:
-
info()for sample information -
binnedData()for the spectrally binned matrices -
accurateData()for the accurate mass information and bin measures for each of the 0.01 amu bins
Bin Metrics
There are a number of metrics that can be computed that allow the assessment of the quality of a given 0.01 amu bin in terms of the accurate m/z mzR::peaks present within its boundaries. These include both purity and centrality.
Purity
Bin purity gives a metric of the spread of accurate m/z mzR::peaks found within a given bin and can be a signal for the presences of multiple real spectral mzR::peaks within a bin.
The purity metric is a value between 0 and 1 with a purity closer to 1 indicating that the accurate m/z present within a bin are found over a narrow region and therefore likely only to be as the result of 1 real mass spectral peak. A reduction in purity could indicate the presence of multiple mzR::peaks present within a bin.
Below shows example density plots of two negative ionisation mode 0.01 amu bins showing high (n133.01) and low (n146.97) purity respectively.
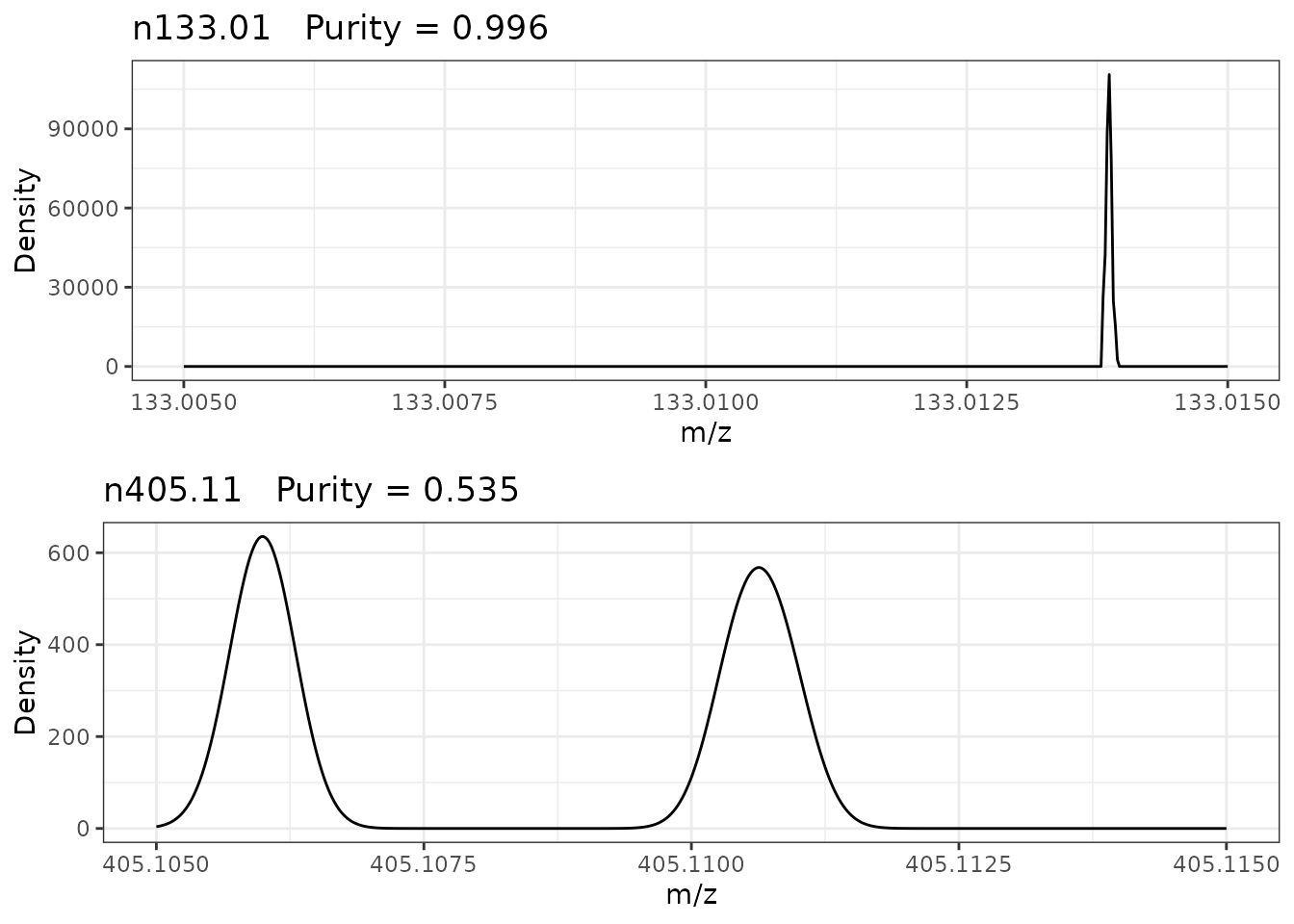
Bin n133.01, that has a purity very close to 1, has only one peak present. Bin n405.11, that has a reduced purity, clearly has two peaks present.
Centrality
Bin centrality gives a metric of how close the mean of the accurate m/z are to the centre of a given bin and can give indication of whether a peak could have been split between the boundary of tow adjacent bins.
The centrality metric is a value between 0 and 1 with a centrality close to 1 indicating that the accurate m/z present within the boundaries of the bin are located close to the centre of the bin. Low centrality would indicate that the accurate m/z present within the bin are found close to the bin boundary and could therefore indicate bin splitting, were an mass spectral peak is split between two adjacent bins.
Below shows example density plots of two negative ionisation mode 0.01 amu bins showing high (n88.04) and low (n104.03) centrality respectively.
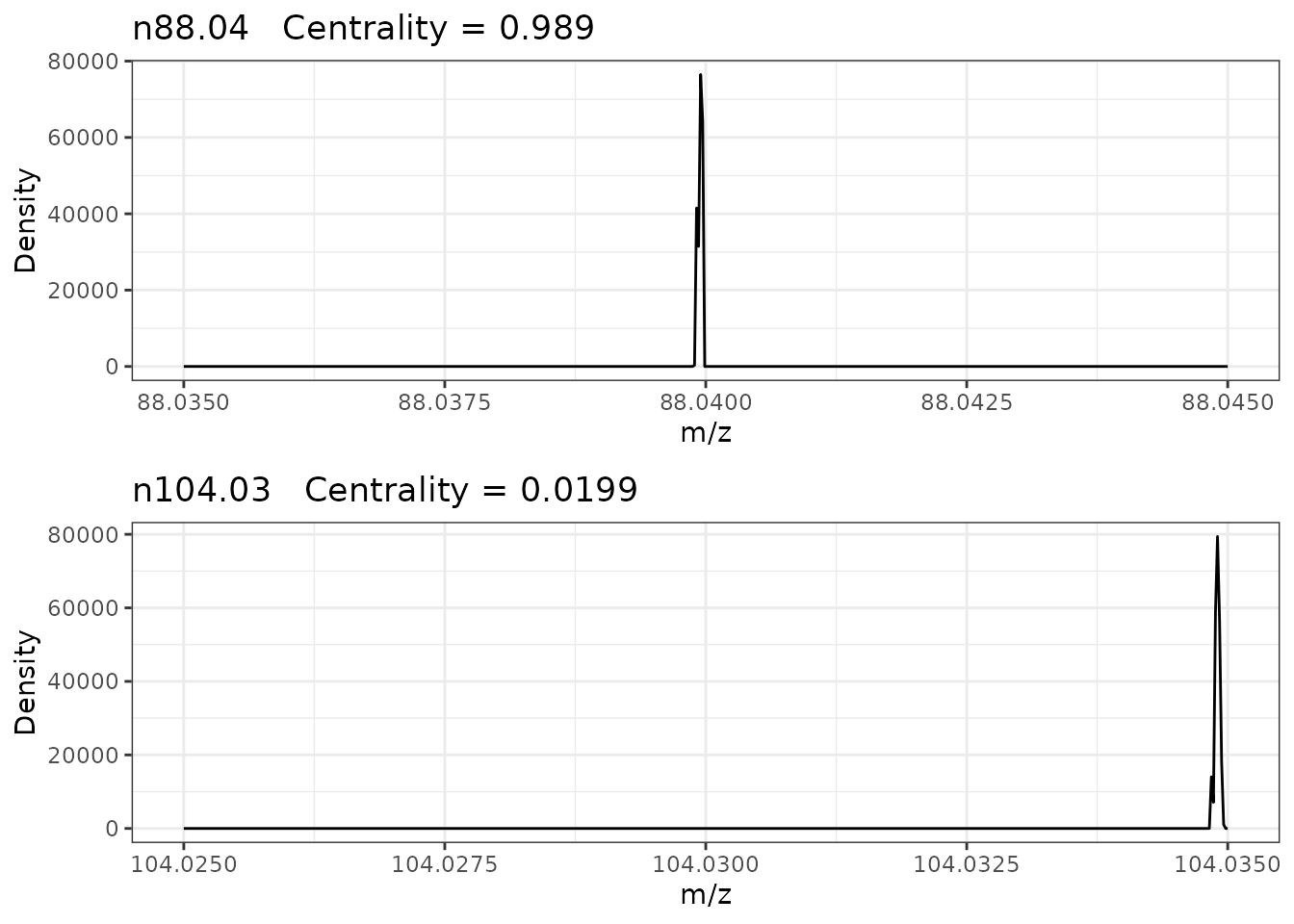
Bin n88.04 has a high centrality with single peak that is located very close to the center of the bin. Whereas bin n104.03 as low centrality with a single peak that is located very close to the upper boundary of the bin and could indicate that it has been split between this bin and bin n104.04.